



Braftovi y Mektovi

Encorafenib está indicado:
• En combinación con Binimetinib en adultos para el
tratamiento del melanoma no resecable o metastásico con
mutación BRAF V600.
• En combinación con Cetuximab, para el tratamiento de
pacientes adultos con cáncer colorrectal metastásico
(CCRm) con mutación BRAF V600E, que han recibido terapia
sistémica previa.
Remite a la Ficha Técnica del medicamento BRAFTOVI®: CIMA: BRAFTOVI 75 MG CAPSULAS DURAS (aemps.es)
PRESENTACIÓN, PRECIO DE VENTA AL PÚBLICO Y CONDICIONES DE FINANCIACIÓN: Caja con 42 cápsulas: PVP: 1.721,42€ y PVP IVA: 1.790,27€. “Comprobar PVP”.
RÉGIMEN Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN: Con receta médica. Diagnóstico Hospitalario.Financiado por el Sistema Nacional de Salud.

Binimetinib está indicado:
• En combinación con Encorafenib en pacientes adultos
para el tratamiento del melanoma no resecable o
metastásico con mutación BRAF V600.
Remite a la Ficha Técnica del medicamento MEKTOVI®: CIMA: MEKTOVI 15 MG COMPRIMIDOS RECUBIERTOS CON PELICULA (aemps.es)
PRESENTACIÓN, PRECIO DE VENTA AL PÚBLICO Y CONDICIONES DE FINANCIACIÓN: Caja de 84 comprimidos: PVP: 3.386,93€ y PVP IVA: 3.522,40€. “Comprobar PVP”.
RÉGIMEN Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN: Con receta médica. Diagnóstico Hospitalario. Financiado por el Sistema Nacional de Salud.





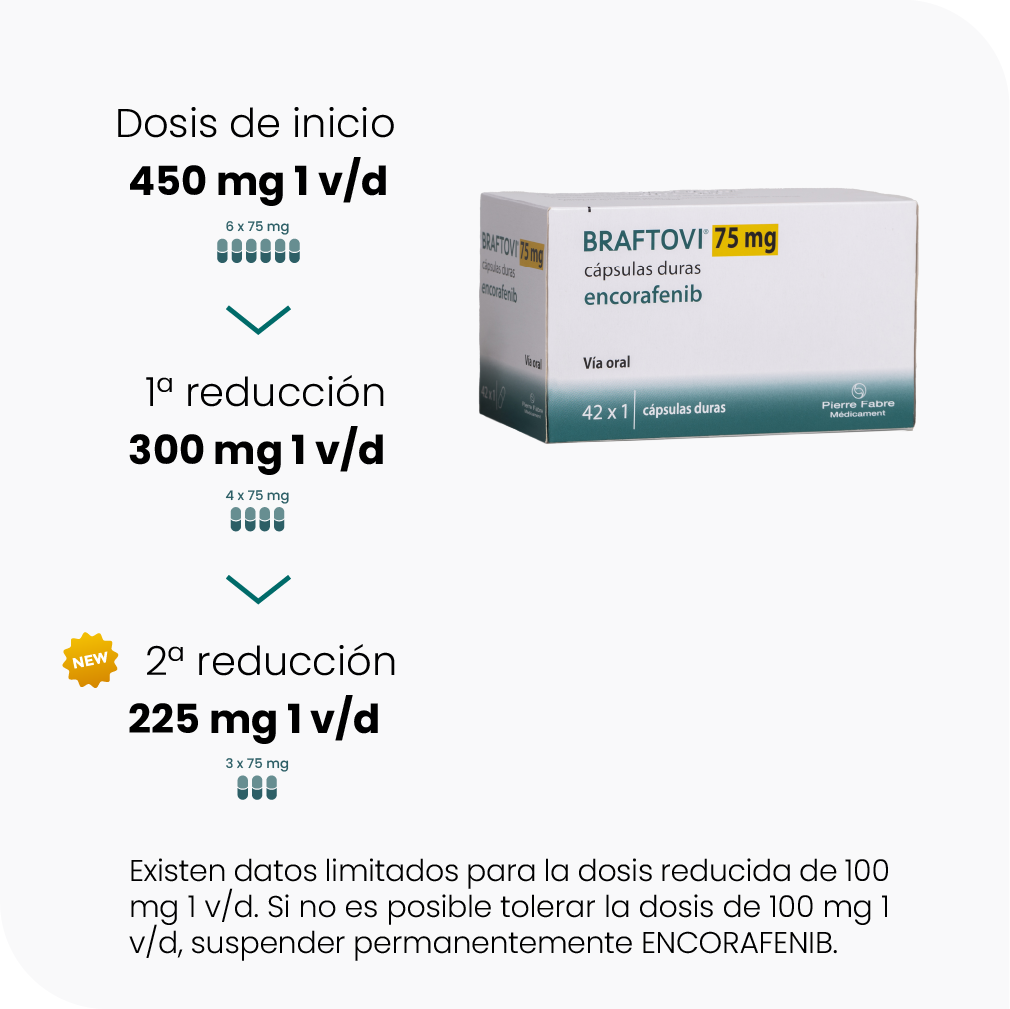
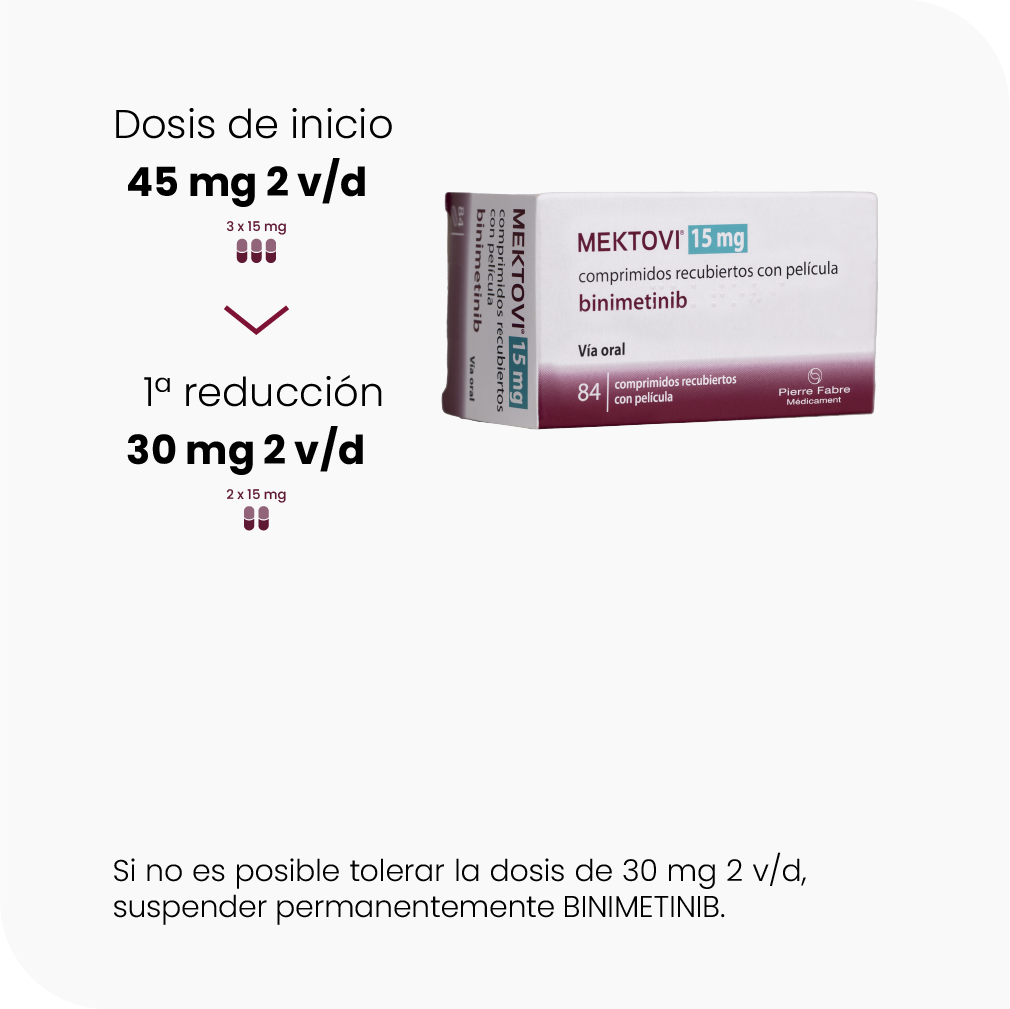
La combinación de BRAFTOVI + MEKTOVI permite una eficacia potente y sostenida(9,10) en el tiempo gracias a las propiedades de PK/PD que pueden traducirse en mejores resultados clínicos y una mayor tolerabilidad para sus pacientes(5).
5. Ascierto PA, Dummer R, Gogas HJ et al. Update on tolerability and overall survival in COLUMBUS: landmark analysis of a randomised Phase 3 trial of encorafenib plus binimetinib vs vemurafenib or encorafenib in patients with BRAFV600-mutant melanoma. Eur J Cancer 2020;126:33–44.
9. Trojaniello C, Festino L, Vanella V et al. Encorafenib in combination with binimetinib for unresectable metastatic melanoma with BRAF mutation. Expert Rev Clin Pharmacol 2019;12(3):259–266.
10. Heinzerling L, Elgentler TK, Fluck M et al. Tolerability of BRAF/MEK inhibitor combinations: adverse event evaluation and management. ESMO Open 2019;4(3):e0004912019;4(3):e000491

Remite a la Ficha Técnica del medicamento BRAFTOVI®: CIMA: BRAFTOVI 75 MG CAPSULAS DURAS (aemps.es)
PRESENTACIÓN, PRECIO DE VENTA AL PÚBLICO Y CONDICIONES DE FINANCIACIÓN: Caja con 42 cápsulas: PVP: 1.721,42€ y PVP IVA: 1.790,27€. “Comprobar PVP”.
RÉGIMEN Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN: Con receta médica. Diagnóstico Hospitalario.Financiado por el Sistema Nacional de Salud.
Remite a la Ficha Técnica del medicamento MEKTOVI®: CIMA: MEKTOVI 15 MG COMPRIMIDOS RECUBIERTOS CON PELICULA (aemps.es)
PRESENTACIÓN, PRECIO DE VENTA AL PÚBLICO Y CONDICIONES DE FINANCIACIÓN: Caja de 84 comprimidos: PVP: 3.386,93€ y PVP IVA: 3.522,40€. “Comprobar PVP”.
RÉGIMEN Y CONDICIONES DE PRESCRIPCIÓN Y DISPENSACIÓN: Con receta médica. Diagnóstico Hospitalario. Financiado por el Sistema Nacional de Salud.